.avif)
Patients
Why SCD is not Getting Government Attention
Patient

Shirley Miller, Patient Advocate/Sickle Cell Disease Consultant
Despite its significant impact, Sickle Cell Disease (SCD) has historically struggled to gain adequate government attention. Several factors contribute to this oversight.
First, the SCD community has long faced disparities in research funding and drug approvals. While I'm disappointed by the limited therapies available for sickle cell patients, it's encouraging to see a few drug companies finally stepping up.
In contrast, many other diseases boast a wide array of treatment options. This highlights the critical need for year-round advocacy and awareness for SCD. Louder voices often lead to greater attention. Compounding the problem are issues within sickle cell care itself, including inequities in access, challenges in obtaining proper care in emergency rooms, and a general lack of equitable treatment.
Second, there's a pressing need to translate stakeholder education into action. While some community-based organizations (CBOs) are highly effective at educating legislators about the impact of SCD, securing consistent funding remains a significant hurdle.
Finally, too many misconceptions about sickle cell disease persist. SCD is a complex condition that presents patients with a wide range of complications. It can be incredibly debilitating, yet its effects are often invisible to the casual observer.
Patients
Transitioning from pediatric to adult care in India
Patient

Girish Dongre, a 21‑year‑old art student living with sickle cell disease (SCD) in India, shares his journey of managing the condition with strong family and healthcare support. Girish has two younger sisters, one of whom also lives with SCD, and their mother—a nurse—plays a central caregiving role. Girish also supports his father, who leads India’s national SCD organization, by editing videos and contributing to advocacy through portrait paintings of inspiring Indian figures.
Reflecting on his early years, Girish says: “I have been living in Nagpur city for the last 15 years with my family. Whenever I get a severe pain crisis, we always prefer the Government Medical College and Hospital (GMC), Nagpur, for treatment. My mother works there as a staff nurse, so it’s more comfortable for me and my family to handle those days. She also takes care of me at home during minor pain crises.” He notes that many doctors in India have limited knowledge of SCD: “Those doctors who are educated about SCD are mostly familiar only with cases in children. Many learned on the spot with patients like me. When a doctor doesn’t know about SCD, my dad would explain to them; he is an enlightened parent.”
Like many children living with SCD in India’s public healthcare system, Girish was cared for by a rotating set of physicians rather than a single dedicated pediatrician. This shifting care environment meant that Girish became accustomed to meeting new doctors frequently—even before adulthood: “I always saw different faces of treating doctors since childhood, so when I transitioned to adult care, I had no fear about that. It’s now just a part of my routine life.” Still, the transition from pediatric to adult care remains a major challenge for SCD patients, including in India. Girish recalls: “I was once hospitalized in an emergency in a hospital in Bombay, not my usual hospital. They kept trying to take blood samples with a needle that was too large for a person having a pain crisis, when blood vessels are contracted, so they finally reverted to the yellow needle, the smaller one for children. After that, they were confused as to how my organs could be in good condition if I have SCD. My dad had to step in and explain that because I receive treatment with hydroxyurea, my organs and hemoglobin are in good condition, but that I still have SCD.”
Girish’s lived experience underscores the urgent need for better-trained healthcare providers, smoother care transitions, and more awareness about adult living with sickle cell disease.
Patients
Transformer l’épreuve en engagement au Niger // Turning the ordeal into commitment in Niger
Patient

Née à Niamey au Niger, Leyla Aïssa Hamidou est diagnostiquée de la drépanocytose à l’âge de deux ans. À l’adolescence, les crises deviennent plus fréquentes, plus intenses, et des complications émergent. Ces atteintes répétées la conduisent plus tard à être prise en charge en France, à l’hôpital Henri Mondor de Créteil, car le manque de ressources et d’infrastructures spécialisées au Niger ne permet pas de traiter des complications aussi sévères. Leyla raconte : « À dix-huit ans, des nécroses ont touché mes hanches et mes épaules. Nous avons dû solliciter l’aide de l’État pour financer mes interventions chirurgicales pour mes deux prothèses de hanche en France, où je suis restée pendant plusieurs semaines. » De retour au Niger, Leyla met tout entre parenthèses pour se concentrer sur sa rééducation et affronter le poids émotionnel de la maladie : « J’ai vraiment raté une partie de ma vie à cause de la maladie et de ses complications. »
Malgré les séquelles physiques et psychologiques persistantes de la drépanocytose — traitements à vie, complications vasculaires et suivi régulier —, Leyla, âgée aujourd’hui de 41 ans, bénéficie au Niger de son traitement de base (hydroxyurée, anticoagulants, acide folique). Elle se rend régulièrement à l’hôpital de Bruxelles, en Belgique, pour des consultations qui lui permettent de prévenir de nouvelles complications. Leyla explique : « La prévention est vraiment clé, que ce soit pour le dépistage mais aussi pour les malades actuels, afin de détecter d’éventuelles complications avant qu’il ne soit trop tard. »
Forte de son expérience et consciente des obstacles au dépistage dans son pays, Leyla fonde en 2023 avec le soutien d’amies et de connaissance l’organisation à but non lucratif DES – Drépanocytose Éducation Santé, afin de sensibiliser la population nigérienne et de promouvoir la prévention. L'ONG collabore avec le ministère de l’éducation pour intégrer la drépanocytose dans les programmes scolaires, et plus récemment avec le ministère de la population de l'action sociale et de la solidarité nationale pour la prise en charge psycho-sociale des patients drépanocytaires. DES – Drépanocytose Éducation Santé organise également des sessions de dépistage directement dans les établissements : grâce à une simple goutte de sang, les élèves peuvent connaître leur statut, avec l’accord de leurs parents. « Il n'y a pas de dépistage néonatal au Niger, si bien que beaucoup de jeunes ignorent leur génotype de la drépanocytose. Ils se marient souvent tôt, juste après leurs études en début de vingtaine ; c’est pourquoi nous les sensibilisons à connaître leur statut. Malheureusement, la seule vraie solution ici reste la prévention », explique-t-elle.
En transformant son expérience personnelle en action collective via DES – Drépanocytose Éducation Santé, Leyla montre que l’information, le dépistage et l’accompagnement peuvent soutenir efficacement une génération de jeunes Nigériens face à la drépanocytose.
English translation:
Born in Niamey, Niger, Leyla Aïssa Hamidou was diagnosed with sickle cell disease at the age of two. During adolescence, crises became more frequent and intense, with complications emerging. These repeated health issues eventually led her to seek care in France at Henri Mondor Hospital in Créteil, as Niger lacked the resources and specialized infrastructure to treat such severe complications. Leyla recalls: "At eighteen, necrosis affected my hips and shoulders. We had to seek state assistance to fund my hip replacement surgeries in France, where I stayed for several weeks." Back in Niger, Leyla put everything on hold to focus on rehabilitation and cope with the emotional toll of the disease: "I really missed a part of my life because of the disease and its complications."
Despite the persistent physical and psychological sequelae of sickle cell disease—lifelong treatments, vascular complications, and regular follow-up—Leyla, now 41, receives her basic treatment in Niger (hydroxyurea, anticoagulants, folic acid). She travels regularly to a hospital in Brussels, Belgium, for consultations that help prevent new complications. Leyla explains: "Prevention is truly key, whether for screening or for current patients, to detect potential complications before it's too late."
Drawing on her experience and aware of screening barriers in her country, Leyla founded the nonprofit DES – Drépanocytose Éducation Santé in 2023 with support from friends and acquaintances, to raise awareness among the Nigerien population and promote prevention. The NGO collaborates with the Ministry of Education to integrate sickle cell disease into school curricula and, more recently, with the Ministry of Population, Social Action, and National Solidarity for psychosocial support for sickle cell patients. DES also organizes on-site screening sessions in schools: with just a drop of blood and parental consent, students can learn their status. "There is no neonatal screening in Niger, so many young people are unaware of their sickle cell genotype. They often marry early, right after studies in their early twenties; that's why we raise awareness about knowing their status. Unfortunately, the only real solution here remains prevention," she explains.
By turning her personal experience into collective action through DES – Drépanocytose Éducation Santé, Leyla demonstrates how information, screening, and support can effectively empower a generation of young Nigeriens facing sickle cell disease.
Patients
Patient Quiz, SCD Symptoms
Patient

Question: Scenario: Your friend with SCD says she has a dull pain in her hip that comes and goes. She limps a bit after sitting for too long. What might this be a sign of?
- Hip inflammation
- A bone infection
- Avascular necrosis
- Nerve damage in her lower back
Answer: Sickle cell disease (SCD) can cause red blood cells to become stiff and misshapen, making it easy for them to get stuck in small blood vessels. This can block blood flow and oxygen delivery to tissues — a painful event known as a vaso-occlusive crisis (VOC). These episodes can cause sudden, severe pain almost anywhere in the body.
AVN, or avascular necrosis, happens when repeated VOCs block small blood vessels supplying bone tissue. AVN is especially common in the femoral head, the ball-shaped top of the thigh bone that fits into the hip socket. Over time, this causes bone death and collapse, often starting with deep, dull groin or hip pain that worsens after rest. Most patients eventually require a total hip replacement.
Question: Scenario: Your friend with SCD has been sleeping more than usual and says he gets out of breath walking to class. He’s not sick, and his appetite is normal. What is the likely cause of his fatigue?
- His red blood cells don’t live long enough to carry oxygen efficiently
- His lungs aren’t taking in enough oxygen from the air
- His heart isn’t pumping oxygen fast enough
- His brain uses more oxygen than average
Answer: Sickled red blood cells break down about every 20 days instead of the usual 120. This causes chronic anemia (low red blood cell count). Without enough healthy cells available to carry oxygen, organs experience continuous stress and reduced function.
Question: Which organ that helps fight infection often stops working properly in people with SCD?
- Liver
- Spleen
- Brain
- Pancreas
Answer: People with SCD generally lose spleen function earlier in life, which weakens their immune system. Repeated VOCs in the tiny vessels of the spleen silently cause irreversible damage — a process called “autosplenectomy,” or functional loss of the spleen, which in turn increases the risk of life-threatening bacterial infections, especially sepsis.
Question: A friend who has SCD has been complaining of pain in their legs for the past day, but now they have a mild fever and a dry cough, and they say their chest feels tight. They’re breathing a little faster than usual, but they insist it’s probably just from the pain and want to wait it out. What’s the best next step?
- Let them rest and give them a painkiller
- Encourage them to take deep breaths and drink water
- Call a healthcare provider or go to the ER right away
- Wait an hour to see if their symptoms get worse
Answer: This is acute chest syndrome (ACS), a VOC where sickled cells block small blood vessels in the lungs, cutting off oxygen and causing inflammation. The impact on the lungs is severe, as repeated episodes can lead to permanent scarring. An ACS episode can be triggered by fat particles from damaged bone marrow (fat embolisms) or a lung infection. Symptoms escalate quickly, making prompt medical attention crucial.
Question: Which symptom might indicate that someone with SCD needs urgent medical care?
- Trouble walking or sudden weakness on one side
- Sore throat
- Mild stomach cramps
- Cold hands and feet
Answer: Sickled cells can block blood flow to the brain, causing strokes—even in young children. Sudden weakness, slurred speech, or confusion are warning signs needing immediate treatment.
Question: Scenario: A teenager with SCD has been getting regular blood transfusions for two years. She recently started having upper belly pain and fatigue. Her doctor is worried about iron levels. Why?
- Her liver may be overworked from producing too many red blood cells
- Transfused blood can directly attack the liver
- She’s not eating enough iron-rich foods
- Iron from the transfusions may be accumulating in her liver and becoming toxic
Answer: Frequent transfusions can lead to iron overload, which stresses the liver. Without monitoring and treatment, this can cause liver failure over time. Liver complications are typically less common and less immediate in childhood than spleen issues. Other stressors like opioids, infections, or dehydration during VOCs can further stress the liver and kidneys.
Question: Why can sudden temperature changes be dangerous for people with SCD?
- They lower red blood cell counts immediately
- They disrupt oxygen production in the lungs
- They shock the vascular system, causing vessel constriction/dilation that triggers sickling
- The air pressure shift strains their joints
Answer: The blood vessels either constrict (tighten) or dilate (widen) quickly to adjust to the new temperature. For people with SCD, this quick shift can be dangerous because their red blood cells are already stiff and sticky. These rapid changes in blood flow make it easier for the cells to get stuck and block small vessels, leading to a VOC.
Question: Scenario: Your friend with SCD decides to go for a short swim at a hotel pool while on vacation. The air feels cool, but the water seems warm enough. That evening, he complains of joint pain and feels unusually tired. What has gone wrong?
- He swam too soon after eating
- The cool air triggered vaso-occlusion
- He strained a muscle in the pool
- His blood sugar dropped from the cool air exposure
Answer: Exposure to cold temperatures causes blood vessels to constrict, which slows down blood flow and makes it easier for the sickled cells to block small blood vessels, triggering a VOC.
Sources:
American Society of Hematology. Patient Stories. https://www.hematology.org/education/clinicians/guidelines-and-quality-care/clinical-practice-guidelines/sickle-cell-disease-guidelines/patient-stories. Accessed August 11, 2025.
Texas Children's Hospital. Kennedy's Story: My Sickle Cell Journey. https://www.texaschildrens.org/content/patient-stories/kennedys-story-my-sickle-cell-journey. Accessed August 11, 2025.
The New York Times. Patient Voices: Living With Sickle Cell Disease. https://www.nytimes.com/interactive/2017/well/patient-voices-sicklecell.html. Published 2017. Accessed August 11, 2025.
Weingart, S. IBCC: Sickle Cell Disease and Acute Chest Syndrome. EMCrit Blog. https://emcrit.org/ibcc/sickle-chest/. Published 2020. Accessed August 11, 2025.
Kaul, D., & O'Connell, M. The Spleen. In StatPearls. StatPearls Publishing; 2024. https://www.ncbi.nlm.nih.gov/books/NBK441872/. Accessed August 11, 2025.
Centers for Disease Control and Prevention. Splenic Sequestration. https://www.cdc.gov/sickle-cell/complications/splenic-sequestration.html. Published 2023. Accessed August 11, 2025.
Qureshi, U., Abugroun, A., & Anwer, F. (2023). Splenic sequestration crisis in adults with sickle cell disease: A systematic review. Journal of Hematology, 12(4), 161–170. https://pubmed.ncbi.nlm.nih.gov/37774369/. Accessed August 11, 2025.
- Hip inflammation
- A bone infection
- Avascular necrosis
- Nerve damage in her lower back
Answer: Sickle cell disease (SCD) can cause red blood cells to become stiff and misshapen, making it easy for them to get stuck in small blood vessels. This can block blood flow and oxygen delivery to tissues — a painful event known as a vaso-occlusive crisis (VOC). These episodes can cause sudden, severe pain almost anywhere in the body.
AVN, or avascular necrosis, happens when repeated VOCs block small blood vessels supplying bone tissue. AVN is especially common in the femoral head, the ball-shaped top of the thigh bone that fits into the hip socket. Over time, this causes bone death and collapse, often starting with deep, dull groin or hip pain that worsens after rest. Most patients eventually require a total hip replacement.
- His red blood cells don’t live long enough to carry oxygen efficiently
- His lungs aren’t taking in enough oxygen from the air
- His heart isn’t pumping oxygen fast enough
- His brain uses more oxygen than average
Answer: Sickled red blood cells break down about every 20 days instead of the usual 120. This causes chronic anemia (low red blood cell count). Without enough healthy cells available to carry oxygen, organs experience continuous stress and reduced function.
- Liver
- Spleen
- Brain
- Pancreas
Answer: People with SCD generally lose spleen function earlier in life, which weakens their immune system. Repeated VOCs in the tiny vessels of the spleen silently cause irreversible damage — a process called “autosplenectomy,” or functional loss of the spleen, which in turn increases the risk of life-threatening bacterial infections, especially sepsis.
- Let them rest and give them a painkiller
- Encourage them to take deep breaths and drink water
- Call a healthcare provider or go to the ER right away
- Wait an hour to see if their symptoms get worse
Answer: This is acute chest syndrome (ACS), a VOC where sickled cells block small blood vessels in the lungs, cutting off oxygen and causing inflammation. The impact on the lungs is severe, as repeated episodes can lead to permanent scarring. An ACS episode can be triggered by fat particles from damaged bone marrow (fat embolisms) or a lung infection. Symptoms escalate quickly, making prompt medical attention crucial.
- Trouble walking or sudden weakness on one side
- Sore throat
- Mild stomach cramps
- Cold hands and feet
Answer: Sickled cells can block blood flow to the brain, causing strokes—even in young children. Sudden weakness, slurred speech, or confusion are warning signs needing immediate treatment.
- Her liver may be overworked from producing too many red blood cells
- Transfused blood can directly attack the liver
- She’s not eating enough iron-rich foods
- Iron from the transfusions may be accumulating in her liver and becoming toxic
Answer: Frequent transfusions can lead to iron overload, which stresses the liver. Without monitoring and treatment, this can cause liver failure over time. Liver complications are typically less common and less immediate in childhood than spleen issues. Other stressors like opioids, infections, or dehydration during VOCs can further stress the liver and kidneys.
- They lower red blood cell counts immediately
- They disrupt oxygen production in the lungs
- They shock the vascular system, causing vessel constriction/dilation that triggers sickling
- The air pressure shift strains their joints
The blood vessels either constrict (tighten) or dilate (widen) quickly to adjust to the new temperature. For people with SCD, this quick shift can be dangerous because their red blood cells are already stiff and sticky. These rapid changes in blood flow make it easier for the cells to get stuck and block small vessels, leading to a VOC.
- He swam too soon after eating
- The cool air triggered vaso-occlusion
- He strained a muscle in the pool
- His blood sugar dropped from the cool air exposure
Answer: Exposure to cold temperatures causes blood vessels to constrict, which slows down blood flow and makes it easier for the sickled cells to block small blood vessels, triggering a VOC.
Patients
My Personal SCD Resource Guide
Patient
As a patient who has dealt with SCD for well over a decade, I know firsthand how challenging it can be to find the right information, in the right format, and in the right place. To help newly diagnosed patients navigate the overwhelming amount of information that is out there, I have pulled together this handy list of resources for managing various aspects of the disease.
Pain Management
CDC "Steps to Better Health" Toolkit
Fact sheets on managing acute and chronic SCD pain.
Nationwide Children’s Hospital Sickle Cell – Pain Management
Includes tips for in-home management of pain.
Navigating Bureaucracies
Sickle Cell Medical Advocacy
Resources, tutorials, administrative and legal support designed to help patients navigate healthcare system and get what they need
Sick Cells Health Insurance Toolkit
Helps patients understand insurance terminology and access their benefits
Caregiver Support
Sick Cells – Caregiver Resources
Standalone page with links, information, resources specifically catered to SCD caregivers.
Walking with Warriors
Resources, inspiration, tools. Tailored for SCD caregivers.
SCD Community
OneSCDVoice – Social Wall
Membership-based online community for SCD patients, caregivers, family members to ask questions, share best practices, provide recommendations to each other.
SCD Awareness
NIH – Social Media Resources
Small repository of social media resources to help raise awareness and visibility for SCD
Patients
My Fight for Sickle Cell Care in Switzerland
Patient

David-Zacharie Issom - Professeur assistant, Département d'informatique de gestion (HEG-GE), HES-SO Genève
Living with sickle cell disease (SCD) in Switzerland has presented unique and ongoing challenges. It's a constant struggle to access knowledgeable specialists, find compatible blood for transfusions, and get essential medications reimbursed. The need for antigen-level matching makes finding safe blood especially difficult, and many vital treatments, like hydroxyurea, aren't automatically covered. This often means I have to fight with insurance providers, largely because SCD isn't widely recognized as a major public health issue here.
My care often feels fragmented. SCD impacts all my organs, so I see multiple specialists—nephrology, ophthalmology, cardiology—at least once a year. But coordinating their efforts is a constant battle. General practitioners often get involved late in the process and frequently lack specific SCD expertise, partly because the disease is relatively uncommon in Switzerland. Finding healthcare providers with the skills to manage my condition has been tough from the start. This fragmentation became glaringly obvious when I transitioned from pediatric to adult care. It wasn't a smooth transition; it was more like being dropped, and the care gap was so vast I nearly died many times. I've had to fight for my health, seek out allies, and teach myself how to survive and adapt. It's exhausting for patients to constantly advocate for themselves and even educate their own healthcare providers.
Life with SCD also brings significant social and educational hurdles. Chronic absenteeism from school and work is common, making it hard to maintain any sense of normalcy. Access to social assistance and occupational therapy can be inconsistent, and adapting home and work environments is often necessary just to cope.
While acute care for SCD in Switzerland is excellent, managing chronic care is far more difficult. There are clear political and cultural barriers to recognizing and funding SCD care, even in high-income countries with a lower prevalence of the disease. Though patient perspectives are slowly being included more in research and drug development, it's still not enough.
A pervasive feeling of "always catching up" has defined my life since childhood. This constant urgency impacts my physiological and mental health.
Patients
Managing Sickle Cell Disease from Childhood to Adulthood
Patient
An anonymous SCD patient explains how her experience with managing sickle disease changed over her lifetime.
My journey with sickle cell disease (SCD) started right after birth. I was diagnosed as a newborn through my state's universal screening program. I'm so grateful for that early diagnosis because it gave me immediate access to a pediatric sickle cell clinic. This meant I could get crucial care right away, like a vaccination program and prophylactic antibiotics to help prevent painful crises.
However, the transition from my pediatric clinic to adult care was a significant challenge. A shared experience for many people with SCD, mortality rates tend to increase for those in their 20s and 30s. Even though my pediatric team started counseling me on self-management at age 13, I still struggled with wanting to be a normal teenager. I wanted to take risks and not feel defined by my illness. Many teens with chronic illnesses navigate their own identity alongside their medical needs.
Fortunately, advances in medical care have provided more options for managing SCD as adults. These include new medications that target the root cause of sickling, not just the symptoms. I credit the close collaboration and open communication between my pediatric and adult care providers for getting my disease under control. They helped me understand that managing my health wasn't about giving up my independence, but about empowering me to live a full life.
Today, I feel fully equipped to make informed medical decisions in consultation with my care providers. I've taken charge of my health as an adult and learned how to balance my condition with my life. I've also learned the importance of having a strong support system, including family, friends, and support groups, to help me through the tough times. While there are still challenges, I'm confident in my ability to manage my health and live a vibrant life. This journey has taught me resilience and the importance of advocating for myself within the healthcare system.
Sources:
Treadwell M, Telfair J, Gibson RW, Johnson S, Osunkwo I. Transition from pediatric to adult care in sickle cell disease: Establishing evidence-based practice and directions for research. Am J Hematol. 2011;86(1):116-120. https://pmc.ncbi.nlm.nih.gov/articles/PMC5291694/. Accessed August 11, 2025.
Cleveland Clinic. Sickle Cell Disease: Transitioning Patients From Pediatric to Adult Care. Consult QD. https://consultqd.clevelandclinic.org/sickle-cell-disease-transitioning-patients-from-pediatric-to-adult-care. Published February 9, 2024. Accessed August 11, 2025.
Patients
Day in My Life
Patient
An anonymous SCD patient offers a look into a day in their life – from the time they wake up to the time they go to sleep.
My day starts at 6:00 AM, with the sound of my alarm. The first thing I do is go downstairs and take my medication. Adhering to my medication schedule is crucial for managing my condition and preventing painful episodes. I take folic acid everyday and sometimes antibiotics to keep infections at bay. When I have a bad episode, I take pain relievers as well.
After breakfast, which usually includes a healthy mix of fruits, whole grains, and proteins, I prepare for my workday. Nutrition plays a significant role in managing my SCD, eat balanced meals throughout the day. Hydration is also critical -- so I make sure to always carry around a giant Stanley mug with me. I even have reminders on my calendar to refill it!
Work itself can be challenging. I often face fatigue and pain, which makes it difficult to concentrate. My employer is understanding, but there are days when I need to take breaks more frequently or work from home. Managing a pain episode at work involves finding a quiet place to rest, using heat packs, and sometimes taking additional pain medication. It is not easy, but I have learned to listen to my body and take the necessary steps to manage the pain. Most of my coworkers are understanding, but a few are still a bit ignorant.
In the afternoon, I try to stay active with light exercises like stretching or walking. Physical activity helps improve circulation and reduces the risk of complications. We have a courtyard behind our office that I like to take two to three laps around. I must be careful, however, not to overexert myself, as it can trigger a pain crisis.
After I go home, a caregiver arrives to help me with household chores, meal preparation, and sometimes, to just provide emotional support. Having a caregiver is a tremendous help, especially on days when the pain is overwhelming. They remind me to take my medication and ensure that I have everything I need to stay comfortable. My caregiver stays for about 90 minutes each day.
After he leaves, I eat dinner with my family. Dinner is another balanced meal, and I make sure to drink plenty of water. I try to relax and unwind. Most nights, I either watch TV or read.
Before bed, I take my final dose of medication for the day and log in my journal my SCD-related events for the day – crises, emotional challenges, etc. I have found these notes to be helpful when I talk to my provider.
Sources:
Press Office. June's story of sickle cell. ABPI. Published February 29, 2024. Accessed August 11, 2025. https://www.abpi.org.uk/media/blogs/2024/february/june-s-story-of-sickle-cell/
This is what sickle cell looks like: courage, self-belief and survival beyond all odds. What SCD Looks Like. Published November 2022. Accessed August 11, 2025. https://whatscdlookslike.com/2022/11/this-is-what-sickle-cell-looks-like-courage-self-belief-and-survival-beyond-all-odds/
Caregivers
Top 5 Caregiver Challenges
Caregiver

An anonymous SCD caregiver offers insight into the five biggest challenges they face.
Emotional Distress
I feel overwhelmed by seeing my loved one in pain and often hospitalized. The unpredictability of SCD and the recurrent pain crises have led me to become perpetually worried and anxious. I can never have a down moment because my partner might need me. This constant need to be vigilant adds to my emotional burden, causing me to become burnt out, drained, and exhausted. Sometimes I feel helpless about my situation, like there is no solution and no relief in sight. When I do something for myself, I feel guilty about leaving my partner alone or with another caretaker.
Financial Strain
I constantly face financial challenges due to the medical, nutritional and transportation expenses associated with caring for my loved one. It is hard for me to know when I might need to spend money, so I am always trying to save money for a rainy day. In the past, when cashflow was tight, I have asked friends to borrow money, and have even sold off assets, including our second car and my bike. A few times, I put off paying for other household needs just to cover my partner’s care costs.
Isolation & Loneliness
I feel alone most of the time, due to the demanding nature of the care I must regularly provide. I have no time for socializing, and trying to make new ones seems pointless as I will not ever be able to see them. If I make a commitment to hang out with someone, I know there is a good chance that I might break it. As a result, I have withdrawn from my once large circle of friends and family.
Self-Neglect
I used to exercise regularly and eat better, but now I am lucky if I can even go for a daily walk. I regularly neglect my own health because I feel guilty for prioritizing myself over the needs of my partner. My biggest nightmare is if something happened while I was at the gym or running errands. I have no personal interests anymore, except getting through each day. Last year, my immune system was compromised; as a result, I got sick a lot, which made it harder for me to provide effective care. The only time I go to doctor now is if I have a sustained fever – even then, I usually just try to power through.
Lifestyle Disruptions
We try to stick to a schedule, but my partner’s unpredictable pain means I must remain flexible. I do what I can, when I can, but often cancel plans with friends. I wish I could spend more time with my parents, but I am too busy caring for my partner. I frequently call out from work or leave early which my employer dislikes. Even basic tasks like getting an oil change or going to the store are hard because I cannot be away for more than 20-30 minutes, as I am the only one who can quickly respond to my partner’s needs.
Sources:
Amuta, S. The challenges of being a caregiver to my cousin with sickle cell. Sickle Cell Disease News. June 16, 2023. Accessed August 11, 2025. https://sicklecellanemianews.com/columns/challenges-being-caregiver-cousin-sickle-cell/
Blaylark, R. Rae Blaylark: A Story of Hope. Centers for Disease Control and Prevention. May 15, 2024. Accessed August 11, 2025. https://www.cdc.gov/sickle-cell/stories/rae-blaylark.html
Walking with Warriors. Sickle Cell Disease in Our Words. Walking with Warriors. Accessed August 11, 2025. https://www.walkingwithwarriors.com/sickle-cell-disease-in-our-words
Caregivers
My Child's Diagnostic Experience
Caregiver
An anonymous parent of a child who was diagnosed with SCD shares their experience, focusing on the changes taking place in the years immediately post-diagnosis.
Getting a sickle cell diagnosis for my child was frustrating, exhausting, and debilitating — not just for my child, but for me as well. We had no idea that our family carried the trait – all I knew was my child was in pain and suffering. The feeling for me was beyond description; I was very distressed, and so was my whole family. The diagnosis came as a complete shock, immediately changing our lives and our sense of security. It meant adjusting to a constant schedule of medical visits, new medical terms, and the understanding that our family's path would be different.
The early years were defined by intense and constant anxiety. We were in a cycle of managing medication, doctor's appointments, and searching for the subtle signs of a crisis. It felt like we were living on a tightrope, always anticipating the next emergency. Every fever or cry of pain became a source of terror. My parental instincts shifted from simply protecting my child to becoming a dedicated advocate for their health. I saw many doctors who couldn't identify the problem, and some even suggested my child was making it up. It took several emergency room visits and a blood test to finally get a sickle cell diagnosis. The lengthy period of not knowing was deeply demoralizing, and the constant struggle to be heard left me distrustful of the medical system. I realized I had to be a fierce advocate for my child at all times.
Beyond the anxiety came a profound sense of guilt. It's a weight you carry knowing this disease is something you passed on. If I had known earlier that both my husband and I had the SCD gene, we might have had a different conversation about having kids. The feeling of blame is a difficult weight to carry, one that stays with you. You question everything and wonder what could have been different. You replay conversations in your mind and feel the heavy sting of what-if. Guilt is a silent companion, always present in moments of my child's pain.
Thank God our other children were spared, but even that brings its own strain, as you try to balance the immense care needs of one child with the attention due to the others. The emotional, social, and psychological weight is immense, but it's a challenge you learn to navigate, one crisis at a time. Responsibility becomes a part of who you are, shaping every decision and every day.
Sources:
Costa, C. P. B., Martins, F. B. S., Ribeiro, C. S., Vasconcelos, S. G., & Santos, M. P. (2017). The Lived Experience of Parents of Children with Sickle Cell Disease: A Qualitative Study. The Open Journal of Nursing, 9(8), 1-8. https://www.scirp.org/journal/paperinformation?paperid=80741
Santos, F. C. B., Pimentel, R. K., Santos, M. P., & Pimentel, R. B. (2020). Family management of children who experience sickle cell disease: a qualitative study. Revista Brasileira de Enfermagem, 73(4), e20190865. https://www.scielo.br/j/reben/a/5KpFYkKjLxLBtXnSszTmYXq/
Healthcare Professionals
Why Many Countries and Communities Struggle with Early Diagnosis
Provider

Dr. Isaac Odame
Medical Director of the Global Sickle Cell Disease Network, Centre for Global Child Health at The Hospital for Sick Children (SickKids), Professor of Pediatrics, University of Toronto
It truly perplexes me why we continue to falter in the early diagnosis and detection of sickle cell disease, despite having the solutions at our fingertips. As I've often said, all we need are a few drops of blood. We possess the technology to create tools that are not only rapid, yielding results in minutes, but also accessible at the most basic levels of healthcare. This should be our reality, just as rapid malaria tests have transformed that landscape.
Yet, here we are, still struggling with robust newborn screening and early infant diagnosis programs. In many resource-limited settings, there is still a critical lack of comprehensive laboratory infrastructure and trained personnel needed for systematic screening. Even when a diagnosis is made, challenges persist in tracking affected infants and linking them to long-term care, often due to factors like short maternity stays and inadequate follow-up systems. Sociocultural factors, including stigma surrounding the disease and insufficient public awareness, also play a significant role in hindering early detection and comprehensive care.
But our main barrier isn't a lack of technological prowess; we absolutely have the capacity to make this testing available at every point of care. Indeed, we have the tools; what we desperately need is a unified, competitive, and favorable global plan. Manufacturers, given sufficient demand—and the demand is undeniably present, consider a million births annually in Ghana alone—could produce these kits for less than half their current exorbitant cost of $3-4 USD per test. This price point remains a monumental hurdle for countless health systems, particularly when faced with other pressing public health priorities.
The core issue, as I see it, is a profound lack of concerted global effort to address these multifaceted challenges. This isn't an insurmountable problem. If we can overcome this inertia and collaborate effectively to ensure affordable diagnostics and robust follow-up care, the widespread early diagnosis of sickle cell disease can be solved in no time."
Healthcare Professionals
What is Sickle Cell Disease?
Provider

Sickle Cell Disease (SCD) is an inherited blood disorder that affects red blood cells. Normal red blood cells are round and flexible, which lets them travel through small blood vessels to deliver oxygen to all parts of the body.
Healthcare Professionals
What is Hydroxyurea?
Pharmacist
I'm often asked about the best treatment for sickle cell disease (SCD), and my answer is almost always the same: hydroxyurea. A standard part of SCD care, this medication is one of the most frequently requested prescriptions I see in my pharmacy.
For people with SCD, red blood cells can become stiff, C-shaped, and sticky. This causes them to get stuck in blood vessels, which can be incredibly painful and damage organs. Hydroxyurea helps prevent this by making red blood cells smoother, rounder, and more flexible. The easier oxygen flow throughout the body leads to less pain and fewer hospital visits.
Hydroxyurea is highly effective and works for most patients, including children as young as nine months old. We frequently prescribe it to children because it can help them live longer, healthier lives with less of the physical and emotional trauma that often comes with SCD.
Although it’s not a cure, hydroxyurea has been a lifesaver for many patients. It's often a preferred treatment over other options like blood transfusions and stem cell transplants, which can cause short- and long-term complications. While hydroxyurea has been used since the 1980s, it wasn't widely approved for SCD until the late 1990s.
Going forward, a major goal is to make hydroxyurea more accessible, especially for children in low-income communities and countries. Early access to this therapy can help kids avoid the debilitating pain and complications that are typically associated with SCD.
Sources:
Hydroxyurea for People With Sickle Cell Disease. KidsHealth. https://kidshealth.org/en/parents/hydroxyurea.html. Published 2022. Accessed August 11, 2025.
Brandow, A. M., & Panepinto, J. A. (2015). Hydroxyurea therapy for sickle cell anemia. Expert Opinion on Drug Safety, 14(7), 1145–1152. https://pmc.ncbi.nlm.nih.gov/articles/PMC5868345/
Healthcare Professionals
Pain Management Body Graph
Provider

When you have SCD, pain comes mostly in the joints, because sickle-shaped red blood cells block blood flow in small vessels, which can restrict oxygen and cause inflammation and tissue damage.
Providers may consider different approaches for pain management, depending on the patient, their medical history, and the kind of pain.
For day-to-day pain, providers may recommend cognitive and behavioral pain management techniques, as well as low risk activities like yoga, massage, and nerve stimulation. In some cases, virtual reality programs have also been successful in helping to alleviate patient symptoms.
Providers may prescribe additional medications and therapies, including SNRIs, to patients struggling with chronic pain. For acute episodes, they may also recommend opioid therapy and prescribe an additional dose of NSAIDs. In hospital settings, providers could deploy alternate remedies, including a ketamine infusion or even regional anesthesia.
Sources:
McClish DK, Smith WR, Dahman BA, et al. Pain Site Frequency and Location in Sickle Cell Disease: the PiSCES Project. Pain. 2009;145(1-2):246-251. https://pmc.ncbi.nlm.nih.gov/articles/PMC2771372/. Accessed August 11, 2025.
Mayo Clinic. Sickle cell anemia: Symptoms and causes. https://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia/symptoms-causes/syc-20355876. Published 2023. Accessed August 11, 2025.
Brandow AM, Carroll CP, Creary S, et al. American Society of Hematology 2020 guidelines for sickle cell disease: management of acute and chronic pain. Blood Adv.
Healthcare Professionals
Helping SCD Patients Manage Medications and Symptoms
Pharmacist

Patients with sickle cell disease (SCD) are regulars at my pharmacy, and I consider it part of my job to keep track of what they need and offer help wherever I can. While technically curable through a bone marrow transplant, SCD for most people is a lifelong condition that can only be managed with medication, which is why a strong relationship with their pharmacist is so important.
A key medication we provide is hydroxyurea, which significantly reduces painful episodes. It’s effective for patients of all ages, but it has to be taken consistently and monitored closely. I help educate patients about their medication, keep a close eye on their lab results, and work with their healthcare providers to adjust doses when necessary. Another critical part of my role is making sure patients are up-to-date on their vaccines and screenings. We want to avoid preventable illnesses that could further complicate an already painful disease.
Because SCD is lifelong, my patients come from all age groups. For children, I work closely with caregivers since the young patients may not fully understand what's happening—after all, what kid likes taking medicine? The kids I work with are resilient, though; they learn a lot quickly and are incredibly brave. By the time they're teenagers, they're usually used to the routine and start taking responsibility for their own care, which can have its ups and downs. I try to meet them where they are and be a trusted, non-judgmental resource when things get tricky.
My older patients have been living with SCD for a long time and have sometimes been let down by the healthcare system. While introducing them to new treatments is important for improving their care, many live by the adage, "if it ain't broke, don't fix it." We do our best to find a balance and respect their experiences.
Ultimately, managing SCD is a joint effort between pharmacists, doctors, nurses, and sometimes social workers. Communication has to be seamless to prevent patients from falling through the cracks, which can lead to too many emergencies for my liking. By working together, we can keep things on track and help people feel better and more confident about their care.
Sources:
Weaver SB, Nonyel NP, Rungkitwattanakul D. Roles of Pharmacists in the Management of Sickle Cell Disease in Adults: A Narrative Review. J Pharm Technol. 2024;40(2):92-99. doi:10.1177/87551225231222437.
Han J, Bhat S, Gowhari M, Gordeuk VR, Saraf SL. Impact of a Clinical Pharmacy Service on the Management of Patients in a Sickle Cell Disease Outpatient Center. Pharmacotherapy. 2016;36(11):1166-1172. doi:10.1002/phar.1834.
Han J, Kemiki O, Hsu LL, Rivers AE. Adverse Reactions to Pneumococcal Vaccine in Pediatric and Adolescent Patients with Sickle Cell Disease. Pharmacotherapy. 2015;35(7):696-700. doi:10.1002/phar.1607.
Weaver SB, Nonyel NP, Rungkitwattanakul D. Clinical consultation: The pharmacist’s role in hydroxyurea management for sickle cell disease. Am J Health-Syst Pharm. 2025. In press. doi:10.1093/ajhp/zxaf114.
Healthcare Professionals
Foods to Eat with SCD
Nutritionist

As a nutritionist who advises patients with sickle cell disease, I can tell you that many of them struggle with finding the foods that are best for them. Some of the common challenges these patients struggle with include:
- Ensuring their diets are sufficiently high calorie and getting the right volume and balance of needed nutrients
- Consuming foods with anti-inflammatory properties
- Overcoming difficulties with nutrient absorption
- Resisting pica, the urge to eat things that aren’t food
To support their health, I recommend:
- Staying well hydrated by drinking plenty of water; this helps prevent complications like pain crises and constipation.
- Eating a balanced, nutrient-rich diet with a variety of colorful fruits and vegetables.
- Increasing calorie intake with nutrient-dense foods like avocados, nuts, whole milk dairy, dried fruits, and adding healthy fats such as olive oil and nut butters.
- Eating enough fiber from whole grains, fruits, vegetables, and nuts to reduce constipation.
- Limiting sugary drinks and processed foods, also called “trigger foods”, to reduce inflammation.
- Including foods rich in essential nutrients, as deficiencies may trigger vaso-occlusive crises:
- Folic acid – for healthy red blood cells: dark leafy greens, citrus fruits, beans, lentils, fortified cereals.
- Calcium – for healthy bones: plain yogurt, cow’s milk, tofu, soybeans, kale.
- Magnesium – for reducing the frequency and severity of painful sickle cell episodes: almonds, spinach, soymilk, black beans.
- Zinc – for decreasing pain, risks of infection and ulcers: meat, seafood, nuts, beans, whole grains.
- Vitamin D – for bone health, immunity, and reducing pain: fatty fish (e.g., salmon, sardines), eggs, mushrooms, fortified milk and dairy.
Sources:
NHS Imperial College Healthcare. NHS Trust. Nutrition and sickle cell disease: Information for patients, relatives and carers. https://www.imperial.nhs.uk/-/media/website/patient-information-leaflets/haematology/red-blood-cell-disease/nutrition-and-sickle-cell-disease.pdf?rev=869dd5e85cf24dd69965e1950fbf14d3. Published July 2025. Accessed 11 November 2025.
Sickle Cell Nutrition Academy. Sickle Cell Nutrition Eat Well Guide. https://sicklecellnutritionacademy.com/nutrition-compendium/. Accessed 11 November 2025.
Physicians Committee for Responsible Medicine. Nutrition Guide for Clinicians: Sickle Cell Disease. https://nutritionguide.pcrm.org/nutritionguide/view/Nutrition_Guide_for_Clinicians/1342072/all/Sickle_Cell_Disease. Accessed 11 November 2025.
Healthcare Professionals
Diagnosing, Treating, and Managing SCD in Children
Paediatrician

Early diagnosis is the first and most critical step in helping a child with sickle cell disease (SCD) live a long, healthy life. While SCD can be diagnosed at any age, newborn screening is the most effective approach. One kind of simple test involves taking a few drops of blood from a newborn's heel, which are then analyzed for various conditions, including SCD. If the initial test is positive, follow-up genetic testing, high-performance liquid chromatography (HPLC), or isoelectric focusing can be used to confirm the diagnosis.
Once diagnosed, paediatricians like me can immediately begin working with families to create a disease management plan, which includes approaches for managing pain, preventing infections and dealing with potential complications. Early intervention is especially crucial, as it allows us to improve the child’s health and overall quality of life from the very start. Unfortunately, in many low-resource settings, where SCD is most common, robust newborn screening programs are not widely available. Lack of early detection tragically means that more than half of children with SCD do not live past their fifth birthday.
We work alongside SCD clinics and centers of excellence to help manage the disease and reduce painful complications like vaso-occlusive episodes, or pain crises. A key part of our strategy is to protect our young patients from infection. We recommend an enhanced panel of vaccinations to protect against diseases like meningitis, pneumonia, hepatitis A and B, and influenza as well as penicillin prophylaxis. Additionally, we counsel families on lifestyle strategies like maintaining adequate hydration and avoiding extreme temperatures, which can trigger painful episodes.
For some patients, a daily medication called hydroxyurea can be a game-changer. Starting this drug as early as 9 months of age can significantly reduce the risk for common and serious complications, such as acute chest syndrome.
As their pediatrician, my job is to reassure parents that an SCD diagnosis isn’t a death sentence. I regularly remind them they are not alone, and they have an entire team of providers ready to support them and their child every step of the way.
Sources:
Akpan B, Okike N, Ekuma-Ogbulu C, et al. Sustainability of newborn screening for sickle cell disease in resource-poor countries: a systematic review. PLoS One. 2024;19(6):e0305110. https://pubmed.ncbi.nlm.nih.gov/39241049/. Published 2024. Accessed August 11, 2025.
National Heart, Lung, and Blood Institute. Sickle Cell Disease: Diagnosis. https://www.nhlbi.nih.gov/health/sickle-cell-disease/diagnosis. Published 2022. Accessed August 11, 2025.
Al-Naami K, Croteau S, Zeller B, et al. Acute complications in children with sickle cell disease: Prevention and management. Paediatr Child Health. 2022;27(1):15-28. https://cps.ca/en/documents/position/acute-complications-with-sickle-cell. Published 2022. Accessed August 11, 2025.
ents/position/acute-complications-with-sickle-cell
Healthcare Professionals
Addressing Nutrition Challenges in Sickle Cell Disease Patients
Dietician

Tadala Kolawole, Specialist Renal Dietician, Barts Health NHS Trust, UK
As a dietitian, I see firsthand the immense challenges individuals with sickle cell disease face, especially when it comes to something as fundamental as eating. It can be a constant battle for them, marked by several key difficulties. One of which being perpetual tiredness, a direct consequence of constantly fluctuating hemoglobin levels. This isn't just mild fatigue; it's a profound exhaustion that affects their appetite and the energy needed for meal preparation. Furthermore, when every bit of effort feels like too much, simply getting a meal together becomes an "extra thing to do," and often, it just doesn't happen.
My primary goal is to ensure they always have a lifeline to proper nutrition, even on their toughest days. I frequently remind my patients, "You need to eat. You need to have something." Sometimes they go for extended periods without adequate nutrition, particularly when they're in and out of the hospital battling frequent crises. During these times, the idea of a full meal can be overwhelming or sometimes not enough to meet the increased metabolic demands the crisis demands.
That's why I often recommend batch cooking and freezing meals or oral nutritional supplement drinks readily available. These aren't always necessary, but they become invaluable during a crisis or when eating feels impossible. It's often much easier for patients to drink these supplements, which contain the essential components of a full meal, than to chew and swallow solid food. While not a complete meal replacement, they provide crucial nutrients and calories, bridging the gap when their bodies are under immense stress.
Healthcare Professionals
A Different Approach for SCD Patients
Anaesthesiologist
An anonymous anesthesiologist walks through the unique challenges faced by sickle cell disease patients and gives insight into the tailored approaches she uses to address their needs.
As an anesthesiologist, I have a vital role to play in treating patients with Sickle Cell Disease (SCD). Each day, I encounter truly unique cases that require a deep understanding of the complexities associated with SCD and the specific adjustments needed to provide optimal care.
My SCD patients often face pain crises, which are sudden episodes of severe pain caused by the blockage of blood flow due to sickle-shaped cells. Managing these pain crises is a critical aspect of my job. Additionally, due to functional asplenia, SCD patients are more susceptible to infections, which can complicate surgical procedures and anesthesia. Chronic hemolysis and vaso-occlusion can lead to damage in organs such as the liver, kidneys, and lungs, requiring careful monitoring and management. The destruction of red blood cells leads to anemia, which can complicate anesthesia and surgery.
To address these challenges, I make several specific adjustments in my approach to anesthesia. I conduct thorough preoperative assessments to understand the patient's medical history, current medications, and the severity of their SCD. This helps in planning the anesthesia and anticipating potential complications. Ensuring adequate hydration and oxygenation is crucial, so I administer intravenous fluids and supplemental oxygen to prevent sickling of red blood cells and maintain proper tissue perfusion. As effective pain management is essential, I use a combination of opioid and non-opioid analgesics, regional anesthesia techniques, and patient-controlled analgesia (PCA) during the postoperative period to manage pain while minimizing side effects.
I also take extra precautions to prevent infections, including the use of prophylactic antibiotics and maintaining strict aseptic techniques during procedures. Continuous monitoring of vital signs, oxygen levels, and blood counts is key. I also work closely with a multidisciplinary team, including hematologists and surgeons, to provide comprehensive care. Postoperative care involves close monitoring for complications such as acute chest syndrome, a common and serious complication in SCD patients. I ensure that pain is well-controlled and that the patient is stable before discharge.
My goal in all this is to provide compassionate and effective care, and to help my patients get through their days with as little pain and complication as possible.
Sources:
Sciaini AA. To address this refractory pain, ... ketorolac, and heat packs. Curr Opin Anesthesiol. 2022;34(5):391-398. doi:10.1016/j.coana.2022.06.007. Accessed August 11, 2025. https://www.sciencedirect.com/science/article/pii/S2772594422000991
Advocates
The Need for a Focused Response to Sickle Cell Disease
Advocate

Witnessing the neglect of sickle cell disease (SCD) in global health, I've seen firsthand how the distribution of resources remains uneven despite some progress. To make the biggest global impact, it's crucial for health organizations to strategically target resources toward Sub-Saharan Africa, India, and the Middle East, where the burden is heaviest.
Sub-Saharan Africa: The Heart of the Epidemic
Sub-Saharan Africa is the epicenter of the global SCD crisis, accounting for over 75% of the world's SCD births. In some countries, the prevalence of the sickle cell trait can be as high as 20-30%. This results in hundreds of thousands of newborns with SCD each year, often in under-resourced healthcare systems.
Infant mortality rates for children with SCD in this region are alarmingly high, with estimates suggesting that 50% to 80% of children with SCD die before the age of five. Those who survive face a lifetime of debilitating pain crises, chronic organ damage, and reduced life expectancy. Basic interventions like newborn screening, penicillin prophylaxis, and hydroxyurea treatment are challenging to access or sometimes altogether unavailable.
Investing in Sub-Saharan Africa is both a moral and strategic necessity. By strengthening healthcare infrastructure, training professionals, and ensuring access to affordable treatments, we can dramatically reduce morbidity and mortality.
India: A Silent Crisis
India represents another critical region where SCD is a major public health challenge. The prevalence of the sickle cell gene is high in certain tribal and rural populations across the country. India bears the world's second-largest burden of SCD, with an estimated over one million affected individuals.
Access to diagnosis and care is severely limited, especially in remote areas. Stigma and lack of awareness further exacerbate the challenges. Investing in community-based screening programs, strengthening primary healthcare, and promoting culturally sensitive education are essential steps to address the crisis in India.
The Middle East: A Region Overlooked
The Middle East, particularly countries like Saudi Arabia and Oman, also has a significant burden of SCD, linked to historical malaria exposure.. While some progress has been made, disparities remain. Many individuals still face challenges in accessing timely diagnosis and comprehensive care. Awareness about SCD needs to be enhanced among both the general population and healthcare professionals. Investing in research and fostering regional collaborations can significantly improve care.
A Targeted Approach for Global Impact
Focusing on high-prevalence regions like Sub-Saharan Africa, India, and the Middle East is the most efficient way to address the greatest need and maximize global impact. Prioritizing these areas allows for the development of large-scale programs and infrastructure that can be adapted globally. While we must continue to support individuals with SCD everywhere, a strategic approach offers the best chance to significantly reduce the global burden of the disease.
Sources
1 World Health Organization, Regional Office for Africa. Sickle Cell Disease: The Silent Killer in Africa. World Health Organization, Regional Office for Africa; 2024.
2 World Health Organization, Regional Office for Africa. Sickle Cell Disease: The Silent Killer in Africa. World Health Organization, Regional Office for Africa; 2024.
3 Adigwe OP, Onoja SO, Onavbavba G. A critical review of sickle cell disease burden and challenges in sub-Saharan Africa. J Blood Med. 2023;14:367-376.
4 World Health Organization, Regional Office for Africa. Sickle Cell Disease: The Silent Killer in Africa. May 2024.
5 The Wire. Sickle Cell Anemia: A Significant Public Health Challenge Impacting Many From Marginalised Communities. June 2025.
6 The Wire. Sickle Cell Anemia: A Significant Public Health Challenge Impacting Many From Marginalised Communities. June 2025.
7 Colombatti R, Hegemann I, Medici M, et al. Systematic literature review shows gaps in data on global prevalence and birth prevalence of sickle cell disease and sickle cell trait: call for action to scale up and harmonize data collection. J Clin Med. 2023;12(17):5538.
8Almuhanna AA, Alharbi F, Alsuwaidan A, et al. The Quality of Life of Sickle Cell Disease Patients in Saudi Arabia 2023. Blood. 2023;142(suppl 1):5310.
Advocates
How You Can Advocate for More SCD Funding
Advocate

In my work as an advocate, people routinely ask me what actions they can take to ensure greater support for sickle cell disease programs and initiatives. Here are the top actions I recommend.
Contact Your Representatives
Reach out to your state legislators, members of Congress and other policymakers to urge them to support increased funding for SCD research, data collection, and comprehensive care programs. Share personal stories and explain the importance of increasing appropriations. Direct staff to resources they can access to learn more.
Contribute to SCD Advocacy Organizations
Directly donate to organizations that are advocating for greater funding for SCD research, treatment, and patient support. Amplify calls to action or other important messages these groups put out. Volunteer your time to support advocacy efforts.
Promote SCD Awareness
Use your own social media platforms to increase awareness about SCD, its impact and the need for greater investments in research. Write op-eds and letters to editor in local media outlets to drive broader support increased funding and education. Educate friends within your own network.
Fundraise for SCD
Organize or take part in charity events, online fundraisers, or donation drives that support SCD research and patient services. Start your own donation page, using integrations already available on social media platforms. Leverage awareness days or other milestones as a means of soliciting donations from your network.
Sources:
Hildenbrand AK. How to Support Someone With Sickle Cell Disease. Nemours KidsHealth. October 2023. Accessed August 11, 2025. https://kidshealth.org/en/teens/sickle-cell-friend.html
Nicol-Wilson D. How to support a friend with sickle cell disease. Sickle Cell Anemia News. May 27, 2024. Accessed August 11, 2025. https://sicklecellanemianews.com/columns/how-to-support-friend-sickle-cell-disease/
Advocates
Fostering Patient Engagement and Policymakers Awareness in India
Advocate

Gautam Dongre - Secretary, National Alliance of Sickle Cell Organizations NASCO India; Board Members, Global Alliance of Sickle Cell Disease Organization
Gautam Dongre, father of Girish and another daughter living with sickle cell disease (SCD), is a leading advocate and patient organization leader in India. For over six years, he and his family have worked to raise awareness, promote education, and improve care for people living with SCD. Together, they helped establish the Alliance of SCD Organizations in India (NASCO), now active across 32 states—17 with a high SCD burden, and organized representation in 11 of them. He also serves as a board member of the Global Alliance of Sickle Cell Organizations (GADSCO).
Their Alliance is recognized by the Government of India as a key partner in national sensitization efforts and contributed to the creation of the SCD Elimination Commission launched two years ago. Gautam is regularly invited to represent the patient community in national forums and health dialogues. The organization conducts webinars, seminars, and state- and national-level roundtables that convene healthcare professionals, policymakers, and patient voices. “We discuss problems openly,” he says, “so that experts can suggest solutions and policymakers can take them home.”
Education and communication are at the heart of their efforts. Through a YouTube channel in regional languages, they help families and healthcare providers understand daily disease management—covering topics such as hydroxyurea (HU) use, nutrition, hydration, and avoiding extreme weather. “People who listen to our videos said that they live better lives—they eat better, know how to use HU, and manage pain crises,” Gautam notes.
He stresses that while government action is growing, awareness among healthcare providers remains inconsistent. “Early diagnosis is very important, but if healthcare professionals are not aware, it doesn’t happen,” he explains. To help close this gap, his organization advocates for training programs and tracks HU availability to alert policymakers where shortages occur.
Social stigma remains another challenge. Because each state has its own language and culture, “stigma changes from one region to another—some think it’s due to food habits, some see it as a curse from the Goddess, while others have moved past it.” Marriageability remains a common concern for families, but Gautam emphasizes that SCD is purely genetic: “We explain this again and again—it will take time, but awareness is growing.”
Despite progress, he estimates that “only about 2 to 5 percent of patients in India are truly settled—meaning they are properly diagnosed, treated, and supported.” Still, through education, advocacy, and peer networks, Gautam remains hopeful: “Every person with SCD can do very well in their activities. We try to show that there is life, creativity, and purpose beyond the boundaries of this disease.”
Advocates
Economic Reasons SCD Remains Untreated
Advocate
Sometimes policymakers who do not fully understand sickle cell disease will ask me questions like “If the treatments are there, why don’t the patients just get them?” As an advocate, my job is to explain the various economic barriers patients may face in their effort to treat SCD. By doing so, I am generally able to convince them that greater action is needed.
Here is a glimpse of the arguments and facts I show them when making my case:
- Unaffordable Treatments
Many patients forgo treatment due to high costs. Out-of-pocket costs are nearly four times higher than those without the disease, sometimes amounting to 5–10% of annual income. Over a lifetime, individuals with SCD pay about $44,000 in OOP medical expenses. Additionally, in the United States, one study found that 20% of SCD patients have no insurance coverage at all. Moreover, treatments like hydroxyurea are often priced out of reach for many patients globally, especially in low-income regions.
- Coverage Barriers
Insurers can deny or delay access to novel therapies for SCD patients and can make the process of accessing traditional therapies more cumbersome. SCD patients on Medicaid also can face difficulty in accessing medication, specialist visits and advanced therapies.
- Logistical Roadblocks
Some therapies require patients to spend one to two months away from home, which is not feasible for many families. SCD patients must travel long distances to specialized centers, incurring additional costs for transportation, lodging, and lost wages from missed work."
- Insufficient Systemic Investment
Inadequate reimbursement mechanisms for SCD also disincentivize health systems from providing high-quality care for patients.
Sources
Johnson KM, Jiao B, Ramsey SD, et al. Lifetime medical costs attributable to sickle cell disease among nonelderly individuals with commercial insurance. Blood Adv. 2023;7(3):365-374. https://ashpublications.org/bloodadvances/article/7/3/365/485129/Lifetime-medical-costs-attributable-to-sickle-cell
Maia M. Lifetime Medical Costs for SCD Total $1.7M, Partly Out of Pocket: Study. Sickle Cell Anemia News. Published May 19, 2022. Accessed July 30, 2025. https://sicklecellanemianews.com/news/scd-patients-lifetime-medical-costs-come-1-7-million-partly-out-pocket/
Johnson KM, Jiao B, Ramsey SD, et al. Lifetime medical costs attributable to sickle cell disease among nonelderly individuals with commercial insurance. Blood Adv. 2023;7(3):365-374. https://ashpublications.org/bloodadvances/article/7/3/365/485129/Lifetime-medical-costs-attributable-to-sickle-cell
Gilbert SK. The Health Insurance Plight of Patients With Sickle Cell Disease. J Natl Med Assoc. 1986 Jul;78(7):663-664, 665. https://pmc.ncbi.nlm.nih.gov/articles/PMC2571400/
Clinton Health Access Initiative. Breaking through barriers: How US$1 tests and affordable treatment are transforming sickle cell disease care. Clinton Health Access Initiative Blog. Published June 3, 2025. Accessed July 30, 2025. https://www.clintonhealthaccess.org/blog/breaking-through-barriers-how-us1-tests-and-affordable-treatment-are-transforming-sickle-cell-disease-care/
Klarer J. 5 barriers to access for sickle cell disease gene therapies. Norstella. Published May 24, 2023. Accessed July 30, 2025. https://www.norstella.com/5-barriers-access-sickle-cell-disease-gene-therapies/
2023 Medicaid Landscape and Access Review for Sickle Cell Disease. Sick Cells; 2023. https://sickcells.org/wp-content/uploads/2024/04/2023-Medicaid-Access-and-Landscape-Review-for-SCD.pdf
Perspectives of individuals with sickle cell disease on barriers to care. PLoS ONE. 2022;17(3):e0265342 https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0265342
Grismore C, Roberts LR, Lister ZD, et al. Barriers to Care for Adults With Sickle Cell Disease: A Qualitative Descriptive Study. Health Expect. 2025;28(3):e70310. https://pmc.ncbi.nlm.nih.gov/articles/PMC12102609/#hex70310-bib-0031
Advocates
Comment Ma Fille A-t-elle été Diagnostiquée / How My Daughter Was Diagnosed
Advocate

Source : Dr Constant Vodouhé, PhD Pharmacology and Cellular Biology, Clinical trials.
President of Community (Patient) Organization, DORYS, Strasbourg – France, Dr Constant Vodouhê, docteur en pharmacologie et biologie cellulaire, essais cliniques. Président de l'organisation communautaire (de patients) DORYS, Strasbourg – France
« Dorys était malade, donc nous nous sommes rendus à l'hôpital, elle pleurait beaucoup, vraiment beaucoup, et depuis plusieurs jours. Une de nos amies nous a dit : « la manière dont cette enfant pleure, elle a mal, ce n'est pas un caprice, elle a mal. » On lui fait des examens, elle avait les pieds et les mains enflés, une des caractéristiques de la drépanocytose, mais on ne le savait pas à ce moment-là. A l'hôpital, ils disent que comme c'est un nourrisson, la fracture n’est pas toujours visible avec la radio, qu’elle est sûrement mal tombée et qu’elle s'est cassée le bras. Nous soupçonnions même que cela s’était produit lorsqu’elle s’amusait avec sa grande sœur et qu’elle ne l’avait pas signalé. Ils lui ont mis des compresses alcoolisées.
Quelques temps après, le même problème se reproduit. Nous nous rendons de nouveau à l’hôpital - on se dit que cette fois, on ne va pas mettre une compresse d’alcool. On y retourne une troisième fois, même chose… on commence par nous soupçonner de maltraitance d'enfant. Finalement, un interne nous demanda si on a un problème de sang dans la famille. On répond que oui, et qu’on a fait le test à la naissance pour savoir si l'enfant serait malade, mais que nous n’avons eu aucun retour concernant les résultats du test - et que nous avons donc considéré qu’il n’y avait aucun problème. Cependant, il y a une eu une faille dans le système car nous sommes remontés jusqu'au prélèvement sur du papier buvard - l’analyse a bien été faite et il n’y a pas eu de suite, nous n’avons jamais reçu les résultats. Finalement, le téléphone sonne à la maison, ma femme décroche. On lui annonce que notre enfant a la drépanocytose - c'était comme annoncer la mort. A partir de ce moment-là on a décidé de se battre. »
English translation :
« [Our daughter] Dorys was sick and on our way to the hospital, she cried so, so much and for many days. One of our friends said, “How she’s crying shows she is in pain, it’s not a whim, she’s in pain.” So, we went to the hospital, and she took the tests - her hands and feet were swollen. She showed the characteristics of sickle cell disease, but we didn’t know at the moment. At the hospital, they said that since she was an infant, a fracture does not always show up on an X-ray—that she must have fallen badly and broken her arm. We even suspected it might have happened while she was playing with her older sister and that she hadn’t said anything. They applied alcohol compresses to it.
Some time after, the same problem came back. We went back to the hospital—we told ourselves that this time we wouldn’t just put an alcohol compress on it. We returned a third time, same thing—and they started to suspect us of child abuse. Finally, a medical intern asked if there was a blood problem in the family. We replied that yes, and that we had done the test at birth to see if the child was sick, but we had never received any feedback about the test results—and so we had assumed that there was no problem. However, there was a failure in the system, because when we traced it back to the blood sample taken on filter paper, the test had indeed been done, but there was no follow-up; we never got the results. Eventually, the phone rang at home, my wife answered. They told her our child had sickle cell disease. It was like they gave her a death sentence. And from that moment on, we decided to fight back.”
Advocates
CONSA - Consortium on Newborn Screening in Africa for SCD
Advocate

The Consortium on Newborn Screening in Africa for SCD (CONSA) introduces standard-of-care practices for screening and early intervention therapies (such as antibiotic prophylaxis and immunizations) at participating institutions, screening 10,000 – 20,000 babies per year in each country, and providing clinical follow-up for babies diagnosed with SCD. Hematologists and public health officials participating in the consortium have mobilized networks of screening laboratories, SCD or pediatric hematology clinics, teaching hospitals, universities, and satellite clinics to screen babies and provide clinical services per the consortium protocol. The first newborn screening and clinical networks launched in 2020.
Visit CONSA's website here.
Advocates
Advocating for Sickle Cell Disease within Legislative Bodies
Advocate
A longtime advocate offers her perspective on how to best engage legislators around SCD issues.
Despite being the most common inherited blood disorder in the United States, SCD continues to be overlooked and underfunded in research, patient support, and public health policies. As an SCD advocate, one of my responsibilities is to urge lawmakers – at all levels of government - to prioritize the fight against this devastating disease.
My role as an advocate is not just about raising awareness—it is about turning that awareness into meaningful, systemic change that legislative bodies can drive. And when I speak to legislators, I emphasize three critical areas: robust funding for research, comprehensive patient care, and education and training.
On the issue of research, I explain the reality that federal and state investment in SCD research lags far behind other genetic conditions, but also show them how increased funding can speed up the development of new treatments and, ultimately, a cure.
When it comes to improving infrastructure for patients, I emphasize the importance of enacting policies that ensure individuals with SCD have access to comprehensive healthcare, which includes pain management, specialized treatments, and transition programs to help patients move from pediatric to adult care.
To bolster education and training, I press the need for supporting programs that educate healthcare providers on SCD best practices to reduce disparities in emergency care and improve patient outcomes.
Aside from these policy points, I also pass on stories from patients themselves. Whenever possible, I try to source these experiences from inside their districts – so they know that the information is coming from a constituent. By sharing the stories of families who spend countless days in hospitals, young adults who face work and school interruptions, and parents who feel powerless against systemic barriers, I remind lawmakers that behind these statistics are real lives.
I will be honest with you -- advocating within legislative bodies is challenging, and securing wins can be hard. But I know that continuing to engage legislators is essential for progress. That is why I have dedicated my life to fighting for individuals, families, and communities affected by SCD.
Sources:
DeBaun, M. R. Grassroots Lobbying Efforts Lead to a Win for SCD Patients in Tennessee: An Interview with Michael R. DeBaun, MD, MPH. ASH Clinical News. December 30, 2021. Accessed August 11, 2025. https://ashpublications.org/ashclinicalnews/news/2157/Grassroots-Lobbying-Efforts-Lead-to-a-Win-for-SCD
Advocates
From Pain to Possibility: 4 Priorities to Tackle SCD Globally
Advocate
No child should suffer excruciating pain or die from a disease we already know how to prevent – yet sickle cell disease (SCD) continues to silently claim hundreds of thousands of lives each year. Globally, 7.74 million people live with SCD, with about 400,000 babies born annually. In 2021 alone, 81,000 children under five died from SCD, making it the 12th leading cause of death in young children. The loss of these lives is not inevitable. But to end SCD deaths within our lifetime, four priorities must guide our efforts:
Priority 1: Early Detection & Preventive Care
Universal newborn screening is the frontline defense. A simple heel-prick test at birth enables life-saving interventions like penicillin, routine vaccinations, and transcranial Doppler screening to prevent strokes. Where screening and early care are implemented, child mortality falls by 70–90%. Yet in many low-resource settings, up to 90% of infants with SCD die before age five, often undiagnosed and untreated.
Priority 2: Comprehensive Treatment Access
Once diagnosed, children deserve effective care. Hydroxyurea can reduce pain crises and hospital visits by 50%, and up to 80–87% for some. But today, too many patients still lack access. Hospitals also need clear pain management plans to provide relief faster, improving recovery. When it comes to curative options, bone marrow transplants achieve over 95% survival with matched sibling donors, and emerging gene therapies like CRISPR show promise in eliminating pain crises and transfusion needs. Expanding access to these treatments is vital to breaking the cycle of pain and disability.
Priority 3: Health System Strengthening & Policy
Effective treatment depends on resilient health systems. Training health workers – following models like Baylor–Texas Children’s programs in Africa – equips providers to deliver evidence-based care. Universal health coverage must include SCD treatment to remove financial barriers. National sickle cell registries, like the CDC’s SCDC program, are critical to track disease burden, evaluate interventions, and guide resource allocation.
Priority 4: Community Empowerment & Innovation
Communities are central to this fight. Awareness reduces stigma and encourages timely care-seeking. Investing in research and inclusive clinical trials will accelerate the arrival of affordable, next-generation gene therapies, closing equity gaps across regions and income groups.
A Call to Action
We stand at a decisive moment. With coordinated action on prevention, treatment, systems, and community empowerment, we can end needless suffering and write a new story for SCD – one of hope, healing, and survival
Sources
1 GBD 2021 Sickle Cell Disease Collaborators. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000–2021: a systematic analysis from the Global Burden of Disease Study 2021. Lancet Haematol. 2023;10(8):e585-e599. doi:10.1016/S2352-3026(23)00118-7 https://pmc.ncbi.nlm.nih.gov/articles/PMC10390339/
2 Okeke CO, Okeke C, Asala S, Ofakunrin AOD, Ufelle S, Nnodu OE. Sustainability of newborn screening for sickle cell disease in resource-poor countries: A systematic review. PLoS One. 2024;19(9):e0305110. doi:10.1371/journal.pone.0305110 https://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0305110
3 GBD 2021 Sickle Cell Disease Collaborators. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000–2021: a systematic analysis from the Global Burden of Disease Study 2021. Lancet Haematol. 2023;10(8):e585-e599. doi:10.1016/S2352-3026(23)00118-7 https://pmc.ncbi.nlm.nih.gov/articles/PMC10390339/
4 McGann PT, Hernandez AG, Ware RE. Sickle cell anemia in sub-Saharan Africa: advancing the clinical paradigm through partnerships and research. Blood. 2013;121(20):3863-3869. doi:10.1182/blood-2013-02-453704 https://pubmed.ncbi.nlm.nih.gov/24094945/
5 Petigas L, Seck N, Doupa D, Diagne I, Roth-Kleiner M. Findings supporting neonatal screening for sickle cell disease: an observational study in Senegal. Front Pediatr. 2025;13:1578570. doi:10.3389/fped.2025.1578570 https://www.frontiersin.org/journals/pediatrics/articles/10.3389/fped.2025.1578570/pdf
6 Kanter J, Smith WR. Describing the sickle cell disease adult patient population: a learning healthcare system perspective. J Am Med Dir Assoc. 2011;12(9):640-644. doi:10.1016/j.jamda.2011.02.002 https://www.sciencedirect.com/science/article/pii/S074937971100626X
7 Grosse SD, Boulet SL, Amendah DD, Oyeku CG, Green NS, Gunter J. Administrative data for surveillance and research on hemoglobinopathies. Am J Prev Med. 2011;41(6 Suppl 4):S339-S345. doi:10.1016/j.amepre.2011.09.005 https://pmc.ncbi.nlm.nih.gov/articles/PMC3560868/
8 Al-Agha AE, Al-Ajlan A. Initial results of newborn screening program for sickle cell disease and other hemoglobinopathies, Jeddah, Saudi Arabia. Saudi J Biol Sci. 2017;24(8):1801-1806. doi:10.1016/j.sjbs.2017.05.011 https://www.sciencedirect.com/science/article/pii/S1658387617300444#:~:text=Abstract,investigated%20with%20promising%20initial%20results.
9 Yale Medicine. Gene therapies for sickle cell disease: what you need to know. Yale Medicine website. Published February 28, 2024. Accessed July 30, 2025. https://www.yalemedicine.org/news/gene-therapies-sickle-cell-disease
10 Gray E. Texas Children’s, Baylor revolutionize sickle cell care in Africa. Houston Chronicle. November 29, 2023. Accessed July 30, 2025. https://www.houstonchronicle.com/health/article/texas-childrens-baylor-sickle-cell-africa-20036496.php
11 Centers for Disease Control and Prevention. Sickle Cell Data Collection (SCDC) program. Updated July 15, 2024. Accessed July 30, 2025. https://www.cdc.gov/sickle-cell/scdc/index.html
Researchers
Why SCD Genotypes Matter
Researcher
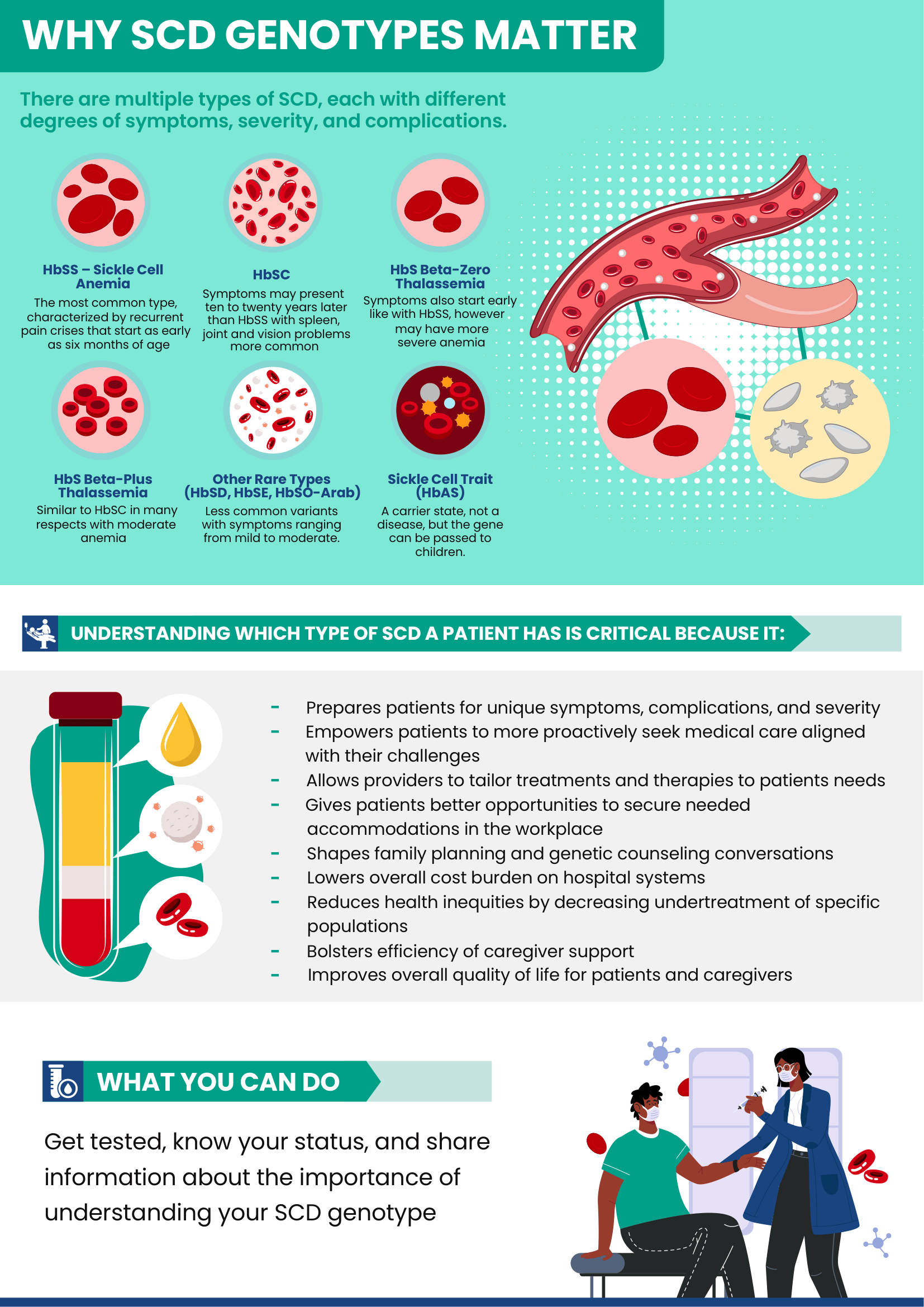
There are multiple types of SCD, each with different degrees of symptoms, severity, and complications.
- HbSS – Sickle Cell Anemia
The most common and severe type, causing frequent pain crises and complications. - HbSC
A milder but still serious form, often with joint pain, fatigue, and vision issues. - HbS Beta-Zero Thalassemia
Almost as severe as HbSS, leading to chronic anemia and pain. - HbS Beta-Plus Thalassemia
Generally milder, with fewer pain episodes and moderate anemia. - Other Rare Types (HbSD, HbSE, HbSO)
Less common variants with symptoms ranging from mild to moderate. - Sickle Cell Trait (HbAS)
A carrier state, not a disease, but the gene can be passed to children.
Understanding which type of SCD a patient has is critical because it:
- Prepares patients for unique symptoms and complications, as well as severity
- Empowers patients to more proactively seek medical care aligned with their challenges
- Allows providers to tailor treatments and therapies to patient’s needs
- Gives patients better opportunities to secure needed accommodations in the workplace
- Shapes family planning and genetic counseling conversations
- Lowers overall cost burden on hospital systems
- Reduces health inequities, by decreasing undertreatment of specific populations
- Bolsters efficiency of caregiver support
- Improves overall quality of life for patients and caregivers
What You Can Do: Get tested, know your status, and share information about the importance of understanding your SCD genotype.
Sources
1 Sampaio, I. G., et al. (2024). "Sickle cell disease: A clinical perspective." Blood, 143(1), 17-26.; Mayo Clinic, Symptoms & causes, 2024; NHS, Sickle cell disease - Carriers, 2025
2 Patel, M., et al. (2023). "Self-efficacy and sickle cell disease knowledge among adolescents." Journal of Pediatric Hematology/Oncology, 45(6), e731-e737.
3 Wang, Y., et al. (2024). "Gene therapy for hemoglobinopathies: current status and future perspectives." Molecular Therapy, 32(1), 1-13; ClinicalTrials.gov, Genetics and Pain Severity in Sickle Cell Disease, 2024).
4 Job Accommodation Network (JAN). (2023). "Accommodation and Compliance Series: Employees with Sickle Cell Disease."
5 Deladem, V. (2024). "Genotype awareness crucial in tackling sickle cell disorder — Expert." The Hope Newspaper.; https://www.ncbi.nlm.nih.gov/sites/books/NBK1377/#:~:text=Genotype%2DPhenotype%20Correlations,occlusive%20events%20%5BSteinberg%202005%5D.
6 Chambers, J. D., O'Day, K., Cangelosi, M. J., Kim, K., Tofarati, T., & Neumann, P. J. (2023). Medical Costs Associated with Sickle Cell Disease by Genotype. JAMA Network Open, 6(11), e2343997.
7 Quinn, C. T. (2024). "Is it time to pay more attention to sickle cell-haemoglobin C disease?" The Lancet Haematology, 11(1), e4-e5.
8 Haj-Hassan, M., et al. (2025). Caregiver and provider perspectives on developmental services for children with sickle cell disease: a mixed methods analysis. Frontiers in Pediatrics, 13, 1530457
9 Nguyen, V. C., et al. (2024). "Factors associated with health-related quality of life in adults with sickle cell disease: a systematic review and meta-analysis." Quality of Life Research, 33(3), 641-655
Researchers
Sickle Cell Disease Trait
Researcher

Sickle cell disease (SCD) is genetic and is caused by a change in a person’s genotype — the combination of alleles inherited from their parents. The most common and severe form, known as sickle cell anemia, occurs when a person inherits two copies of the mutated hemoglobin allele (HbS), one from each parent (HbSS).
Those who inherit only one copy of the mutated HBB allele (HbS) and one normal allele (HbA) have the sickle cell trait (HbAS). Carriers usually do not experience symptoms and live healthy lives but they can pass the HbS allele to their children.
Sources
Powars D. Sickle Cell Disease. Salem Press Encyclopedia of Health. 2024.
Berman JO. Sickle Cell Disease and Genetics. Salem Press Encyclopedia of Health. 2024.
Genetic and Rare Diseases Information Center (GARD). Sickle cell anemia. https://rarediseases.info.nih.gov/diseases/6839/sickle-cell-anemia. Accessed August 11, 2025.
Introduction – Addressing Sickle Cell Disease. In: Sickle Cell Disease. National Center for Biotechnology Information (US); 2023. https://www.ncbi.nlm.nih.gov/books/NBK589600/. Accessed August 11, 2025.
Researchers
More than a Budget Line
Researcher

Source: Prof Clarisse Lopes de Castro Lobo, PhD, Clinical Research Specialist and PHD In Medical Sciences, HEMORIO
Sickle cell disease is one of the most common genetic conditions in Brazil, disproportionately affecting Afro-Brazilian communities. Advances in early diagnosis, access to care, and new therapies have already shown that with the right investment, outcomes can improve dramatically. But progress depends on consistent, targeted research funding — and that’s where we continue to fall short.
As a sickle cell researcher in Brazil, I can attest that one of our most significant hurdles is the inconsistent nature of research funding. It's a common misconception that funding for vital initiatives like newborn screening, and the research supporting it, is straightforward once a budget line item is approved. In reality, our funding processes are far more intricate and, frankly, frustrating.
Our research funding is typically pieced together from both federal and state governments. Health initiatives, by their very nature, are often funded in fragmented ways. However, when it comes to research, this division between federal and state levels complicates project implementation even further. This convoluted structure means that despite a clear and compelling case for investing in SCD research, the actual allocation of funds often hinges on the ability of researchers and advocates to lobby and influence politicians.
Today, to navigate these barriers, SCD researchers in Brazil often find themselves devising workarounds – perhaps by focusing on new medicine trials that attract different funding streams, or by "nesting" smaller projects within larger, more accessible funding pockets. But this isn't sustainable, nor is it efficient.
Instead of relying on these flawed, often politically driven processes, we desperately need more streamlined, dedicated funding for SCD research. This would allow us to bypass bureaucratic obstacles and, most importantly, deliver the tangible results that our patients so urgently need. Investing in this critical research isn't just about scientific advancement; it's about improving and saving lives.
Researchers
Emerging Innovations in Sickle Cell Disease Care: A Transformational Moment
Researcher
Over the past year, significant advances in gene therapy, pharmaceutical treatment, and digital health have reshaped the landscape of sickle cell disease (SCD) care. These developments offer not only new hope for symptom control but also, for some patients, realistic pathways toward long-term remission or cure.
Gene Therapy: A Turning Point in Curative Potential
Two leading gene-based approaches have reached late-stage regulatory review or approval in major markets., One involves gene editing techniques to restore fetal hemoglobin production, thereby preventing the red blood cell sickling that causes most SCD complications. The other strategy delivers a healthy version of the hemoglobin gene into the patient’s cells. Both have shown remarkable clinical outcomes, including the elimination of severe pain crises in most treated individuals during trial follow-ups. These therapies mark a turning point in the pursuit of durable, curative solutions—though access remains limited to specialized treatment centers.
Diagnostic Advances: Faster, Cheaper, Accessible
Point-of-care innovations are revolutionizing SCD diagnosis, especially in low-resource settings:
- Lateral flow strips & paper-based tests – rapid, low-cost field screening.
- Smartphone-based assays – optical/magnetic sensing for quick results.
- HemeChip electrophoresis & microfluidics – portable hemoglobin typing.
These tools enable early detection, reduce childhood mortality, and bridge global diagnostic gaps.
Digital Health: Enhancing Management & Research
Telemedicine platforms, wearables, and smartphone apps allow real-time symptom tracking and provider connection, reducing hospitalizations through early interventions. AI-powered platforms create personalized treatment plans based on medical history and genetics, improving adherence and outcomes. Online education tools and patient communities empower self-management, while digital trial platforms accelerate research and expand participant diversity.
Why These Innovations Matter
Together, these breakthroughs shift SCD care from symptom management to potential cures, integrate rapid affordable diagnostics, and harness digital tools for personalized care. They expand therapeutic options for clinicians, create new markets and partnerships for companies, and provide policymakers with scalable, evidence-based solutions to reduce suffering and mortality worldwide.
Sources
1 U.S. Food and Drug Administration. FDA approves first gene therapies to treat patients with sickle cell disease. FDA.gov. Published 2024. Accessed July 30, 2025. https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease
2 NHS England. NHS rolls out ‘life-changing’ treatment for thousands with sickle cell disease. England.NHS.uk. Published May 3, 2024. Accessed July 30, 2025. https://www.england.nhs.uk/2024/05/nhs-rolls-out-life-changing-treatment-for-thousands-with-sickle-cell-disease/
3 Blood. American Society of Hematology Publications. Accessed July 30, 2025. https://ashpublications.org/blood/article/144/Supplement%201/511/530867
4 The American Journal of Managed Care. Nearly all patients who received exa-cel for SCD are hospital-free after a year, data show. AJMC.com. Accessed July 30, 2025. https://www.ajmc.com/view/nearly-all-patients-who-received-exa-cel-for-scd-are-hospital-free-after-a-year-data-show
5 PMC (PubMed Central). Accessed July 30, 2025. https://pmc.ncbi.nlm.nih.gov/articles/PMC9455601/ https://pmc.ncbi.nlm.nih.gov/articles/PMC9455601/
6 Xcene Innovate. Digital health and sickle cell disease management. XceneInnovate.com. Accessed July 30, 2025. https://xceneinnovate.com/digital-health-sickle-cell-disease-management/
Employees & Employers
How to Effectively Communicate with Your Employer
Employee
An anonymous sickle cell disease patient offers advice on optimizing conversations with employers.
Living with sickle cell disease means constantly balancing your health with the demands of your job. The fear of being seen as unreliable or weak can make it incredibly difficult to open up to your employer about your condition. But having this conversation is a critical step in protecting your well-being and building a successful career. By approaching it with preparation and confidence, you can create a work environment that truly supports you.
Based on my own journey and conversations with others in the SCD community, here are five essential tips to help you navigate this important discussion.
1. Do Your Homework and Know Your Rights
Before you even schedule the meeting, arm yourself with knowledge. First, understand the specifics of your own SCD. How does it typically affect you? What are your common triggers and symptoms? What accommodations have helped you in the past? Next, research your company's policies regarding sick leave, disability, and accommodations. It's also important to familiarize yourself with laws like the Americans with Disabilities Act (ADA), which may protect you and your right to reasonable accommodations. Knowing your rights empowers you and helps you frame your requests within a legal and professional context.
2. Schedule a Proper Meeting
Avoid bringing up your condition in a casual, offhand way. Instead, request a private meeting with your direct manager and, if possible, a representative from Human Resources. This shows that you are serious and want to have a structured, confidential discussion. Prepare an agenda in advance. Having a clear plan—from how you will explain SCD to the specific accommodations you will request—ensures you cover all your points and stay on track.
3. Focus on Solutions
Your employer wants to know that you are a reliable and productive employee. When discussing your challenges, connect your needs directly to your ability to do your job well. For example, instead of simply saying, "I get fatigued and need to rest," you could say, "A flexible schedule would allow me to manage my energy levels and complete my tasks effectively, even on days when I'm feeling low." Presenting your requests as solutions that will help you maintain or even improve your performance is more likely to be met with a positive response.
4. Be Specific and Realistic with Asks
Vague requests can be difficult for an employer to accommodate. Be prepared to offer specific, actionable solutions. Examples of reasonable accommodations might include:
- Flexible Work Schedule: The ability to shift your start and end times or work remotely on certain days.
- Time Off: Understanding for unexpected absences due to pain crises or medical appointments.
- Workplace Environment: A quiet space to rest briefly, access to a small heater in a cold office, or a modified workstation.
- Workload Adjustments: Temporary changes to project deadlines or responsibilities during a health challenge.
Be realistic about what is feasible for your role and your company. A well-defined request is far more likely to be approved.
5. Document Everything
After your meeting, follow up with a concise email summarizing the key points of your conversation and any agreed-upon accommodations. This creates a written record that protects both you and your employer. If you submit a formal accommodation request, keep copies of all correspondence. This is not about being confrontational; it’s about maintaining a clear, professional paper trail that can prevent future misunderstandings.
Sources:
Nicol-Wilson D. Telling My Employer About Sickle Cell Disease Was the Right Decision. Sickle Cell Anemia News. Published October 18, 2021. Accessed August 11, 2025. https://sicklecellanemianews.com/2021/10/18/telling-employer-about-sickle-cell-right-decision/
Sickle Cell Society. Sickle Cell Work and Employment: A Guide for Employers and Employees on Work, Employment and Sickle Cell Disorder (SCD). Published July 2019. Accessed August 11, 2025. https://www.sicklecellsociety.org/wp-content/uploads/2019/07/FINAL_Guide-To-Sickle-Cell-and-Employment-Version_1_2019.pdf
USF Health. Americans with Disabilities Act. Accessed August 11, 2025. https://health.usf.edu/-/media/v3/usf-health/medicine/Orthopaedics/Sickle-Cell/Policies/AmericanswithDisabilitiesAct.ashx
Chiesi USA. Managing Work: Talking to Your Employer. Published 2023. Accessed August 11, 2025. https://resources.chiesiusa.com/OurIronWill/Our_Iron_Will_Managing_Work.pdf
Employees & Employers
How Our Company Supports Employees With Sickle Cell Disease
Employer

As an employer with several employees who have sickle cell disease, we’ve developed a comprehensive approach to ensure they feel supported at work. By creating thoughtful policies and flexible practices, we've discovered that we not only support individuals but also build stronger, more productive teams. Here are some of the practical ways we’ve achieved this:
Awareness & Education
We regularly hold awareness sessions and provide easy-to-read digital resources to educate our team about SCD. Our initiatives have been effective in spreading knowledge, reducing stigma, and fostering a more inclusive and understanding environment for everyone.
Flexible Work Options
To accommodate the unique health needs of our employees with SCD, we’ve implemented flexible hours and remote work options. Leave for health needs is provided without penalty, and our managers are trained to offer support with empathy. The resulting flexibility has been crucial for helping our employees manage their condition while maintaining their productivity.
Workplace Adjustments
We've made specific adjustments to enhance comfort and performance. Our adjustments include ensuring consistent access to water and temperature control, providing ergonomic setups, and allowing for flexible dress codes. We have also customized our attendance tracking and support plans to meet the unique needs of our employees with SCD.
Legal Compliance
Our approach is built on a foundation of legal compliance. We diligently follow disability rights laws like the ADA and FMLA, as well as similar laws in other countries, to ensure our employees with SCD are treated fairly and have the support they need to succeed.
Our inclusive practices have yielded positive results, leading to better employee retention, stronger team morale, and consistent productivity. We firmly believe that supporting employees with SCD isn't just the right thing to do—it builds a more successful workplace for all.
Sources:
Sickle Cell Society. FINAL Guide To Sickle Cell and Employment Version_1_2019. Sickle Cell Society. Published 2019. Accessed August 11, 2025. https://www.sicklecellsociety.org/wp-content/uploads/2019/07/FINAL_Guide-To-Sickle-Cell-and-Employment-Version_1_2019.pdf.
Job Accommodation Network (JAN). Sickle Cell Anemia. AskJAN.org. Accessed August 11, 2025. https://askjan.org/disabilities/Sickle-Cell-Anemia.cfm.
Scott K. How to support employees with sickle cell disorders. HR Magazine. Published May 10, 2024. Accessed August 11, 2025. https://www.hrmagazine.co.uk/content/features/how-to-support-employees-with-sickle-cell-disorders/.
Employees & Employers
How Employees Can Manage SCD in the Workplace
Employee
An anonymous employee and SCD patient shares their perspective on navigating SCD dynamics in the workplace.
Earlier this year, I came across a writeup on the experience of Tasha Hines, who was diagnosed with sickle cell disease (SCD) as a newborn. Today, she works as a successful registered nurse in an internal medicine outpatient clinic – a role that speaks to both her dedication to healthcare and her determination to lead a full and meaningful life while managing a chronic condition.
“Having sickle cell disease is what led me into the healthcare field. Even though my mother didn’t want me to pursue nursing – she thought it was too strenuous and wanted me to have a sit-down job.”
Tasha’s story illustrates what is possible when people like me, living with SCD, have the right tools, resources, and support. For many of us, certain careers may seem out of reach – too demanding, too risky, too rigid. The truth is: the disease doesn’t just attack the body with pain, fatigue, and complications. It also challenges a person’s sense of possibility. But those barriers can be broken. With the right knowledge, support, and workplace environment, people with SCD can build careers that are not just sustainable but deeply fulfilling.
One of the most important and personal decisions I made when starting a new job is when and how to disclose SCD at work. Disclosure is not an obligation; it’s a strategic choice, often made when symptoms begin to affect job performance or when accommodation is necessary. In the U.S., the Americans with Disabilities Act (ADA) protects employees with SCD, recognizing it as a legitimate disability. This legal protection ensures access to reasonable accommodations such as flexible schedules, remote work, additional breaks, or modified duties. Effective disclosure begins with clarity: explain how SCD impacts you at work and propose specific, manageable adjustments. A letter from your physician can strengthen the case and ease communication. But this can vary from country to country, and not everyone is as lucky to have these protections.
Managing SCD on the job requires planning, self-awareness, and support. Simple but vital practices – staying hydrated, pacing yourself, taking short breaks, and knowing when to rest – can make a profound difference. Regular use of treatments like hydroxyurea can drastically reduce pain episodes and hospitalizations. Appointments, therapies, and check-ins are part of life with SCD, and tools like telehealth or early morning visits can keep disruptions minimal. Above all, building a care network - a hematologist, a primary doctor, and mental health professionals - is how I stay strong, both physically and emotionally.
Inclusive workplaces don’t happen by chance; they require education, policy, and culture. Managers should be informed about SCD and understand that its effects can vary greatly between individuals. Organizations can support inclusivity by establishing clear processes for accommodations, incorporating chronic illness awareness into wellness initiatives, and encouraging respectful communication around health disclosures.
By fostering understanding and implementing thoughtful policies, employers and employees alike can contribute to a more inclusive workforce. Individuals with SCD bring unique insights, resilience, and capabilities to their roles. With awareness, mutual respect, and supportive practices, it is possible to create environments where employees with chronic conditions can thrive professionally while maintaining their health and well-being.
Sources:
Centers for Disease Control and Prevention. Persons living with SCD reach adulthood, but life expectancy remains reduced; Real stories from people living with SCD. https://www.cdc.gov/sickle-cell/stories/index.html. Published 2022. Accessed August 11, 2025.
Community for Accredited Online Schools. Guide to sickle cell and employment. https://www.sicklecellsociety.org/wp-content/uploads/2019/07/FINAL_Guide-To-Sickle-Cell-and-Employment-Version_1_2019.pdf. Accessed August 11, 2025.
National Heart, Lung, and Blood Institute. NHLBI SCD fact sheet. https://www.sicklecelldisease.org/links-resources/. Published 2022. Accessed August 11, 2025.
Sickle Cell Disease Association of America. SCDAA C.A.R.E.S. Consortium launches clinical trial awareness. https://www.sicklecelldisease.org/2023/03/15/scdaa-to-promote-clinical-trials/. Published 2023. Accessed August 11, 2025.
Mayo Clinic. Sickle cell anemia: Pain management strategies. https://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia/diagnosis-treatment/drc-20355882. Published 2025. Accessed August 11, 2025.